Ernia Diaframmatica
Chirurgia Pediatrica Catania
C.D.H. - Congenital Diaphragmatic Hernia
Il trattamento dei neonati affetti da Ernia Diaframmatica Congenita (CDH) rappresenta ancora oggi una delle più grandi sfide per il Chirurgo Pediatra, in quanto, nonostante i notevoli miglioramenti nel management medico-chirurgico, il tasso di mortalità permane ancora alto.
Cosa è l'ernia diaframmatica?
L'ernia diaframmatica congenita (CDH- Congenital Diaphragmatic Hernia) è una patologia contraddistinta dalla mancata o incompleta formazione della cupola diaframmatica, con il conseguente passaggio di alcuni visceri addominali nella cavità toracica e con la compressione e dislocazione degli organi toracici. La CDH è una patologia che presenta ancora numerose controversie sia dal punto di vista patogenetico che di trattamento.
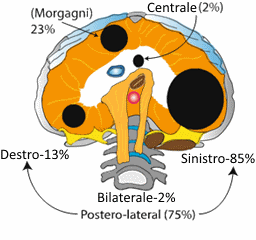
L'eziopatogenesi dell’ernia diaframmatica congenita non è ad oggi chiaramente conosciuta e si presuppone essere multifattoriale. L'ernia posterolaterale, nota come ernia di Bochdalek, è il tipo più comune di CDH (70-75%) con più frequente coinvolgimento del lato sinistro (85%) rispetto al lato destro (13%), mentre la forma bilaterale rappresenta solo il 2% dei casi. Il difetto anteriore, ernia di Morgani, incide per il 23-28%; rara l’ernia centrale (2-7%). Il diametro del difetto è variabile da piccolo (circa 20-30 mm) ad estremamente ampio sino ad avere la completa assenza del diaframma; in rari casi si ha una vera agenesia del diaframma.
Il vero problema della CDH è l'ipoplasia polmonare e l’ipertensione polmonare che compromettono la funzionalità respiratoria e cardiovascolare. Purtroppo non possono essere prognosticate in epoca pre-natale e spesso sono severamente compromesse alla nascita. I polmoni hanno una ridotta superficie di scambio gassoso a causa dell'ipoplasia, in particolare si ha una riduzione dei bronchioli terminali e degli alveoli. Gli alveoli presentano pareti ispessite. Frequentemente possono associarsi altre malformazioni, che incrementano notevolmente la morbidità e la mortalità.
Sebbene sia considerato un difetto “sporadico”, sono stati segnalati alcuni casi familiari; sembrerebbe essere presente un’ereditarietà multifattoriale13, con un rischio di ricorrenza del 2% nel parente di primo grado14. Nonostante l’esatta eziopatogenesi dell’ernia diaframmatica congenita (CDH) rimanga ancora sconosciuta, è sempre più evidente che una componente genetica svolga un ruolo nel suo sviluppo. Cause genetiche sono state riscontrate nel 30% dei casi.
Cosa fare se si sospetta una ernia diaframmatica?
La diagnosi è generalmente posta attraverso l’ecografia prenatale che mostra l’assenza parziale del diaframma e la risalita dei visceri in torace, a questo quadro si associa un polmone più piccolo e iposviluppato. La scelta di proseguire la gravidanza dipende dai genitori che devono essere consigliati e supportati da un team multidisciplinare perché l’evoluzione della malattia dopo la nascita non è purtroppo prevedibile e la prognosi può essere variabile. Certo l’interruzione di gravidanza rappresenta già un’enorme sconfitta, mentre la CDH rappresenta una sfida che può comunque nella maggior parte dei casi essere vinta!! La madre ed il feto andranno indirizzati in un centro adeguato con terapia intensiva neonatale (UTIN) e chirurgia pediatrica, il parto sarà programmato ed il neonato sarà assistito alla nascita dal punto di vista cardiorespiratorio e trasferito in UTIN.
Cosa succede dopo la nascita?
Il neonato sarà monitorato ed assistito in UTIN fino a quando sarà considerato stabile dal punto di vista respiratorio e cardiocircolatorio, solo in quel momento sarà sottoposto ad intervento chirurgico, che potrà avvenire in sala operatoria o in UTIN per i casi più complessi. La prognosi dipenderà dal grado di ipoplasia polmonare, dalla presenza di altre malformazioni associate, se presenti, e dalla risposta individuale del bambino alle cure.
Che tipo di intervento?
L’intervento è generalmente eseguito con chirurgia tradizionale, cioè con un’incisione sottocostale, ed ha lo scopo di ricostruire la parte di diaframma mancante. A volte è possibile chiudere la breccia mediante dei punti , altre volte, per difetti maggiori, potrà essere necessaria l’apposizione di una protesi morbida sul diaframma. È possibile correggere il difetto anche mediante laparoscopia (cioè in maniera mininvasiva, mediante piccole incisioni praticate sull’addome) ma questa metodica è riservata ai casi meno severi che non si presentano alla nascita ma tardivamente.
Tutte le CDH sono severe?
No, la prognosi può essere buona soprattutto nelle patologie non diagnosticate alla nascita ma che si manifestano in bambini più grandi, per questi casi il trattamento è generalmente laparoscopico e non sempre è necessario il ricovero in terapia intensiva.
Quale sarà la qualità di vita del bambino?
La qualità di vita è in genere buona, tutto dipende dalla presenza di malformazioni associate o dal grado di ipoplasia polmonare per la quale potrebbe essere necessaria ossigeno terapia.

Navigazione